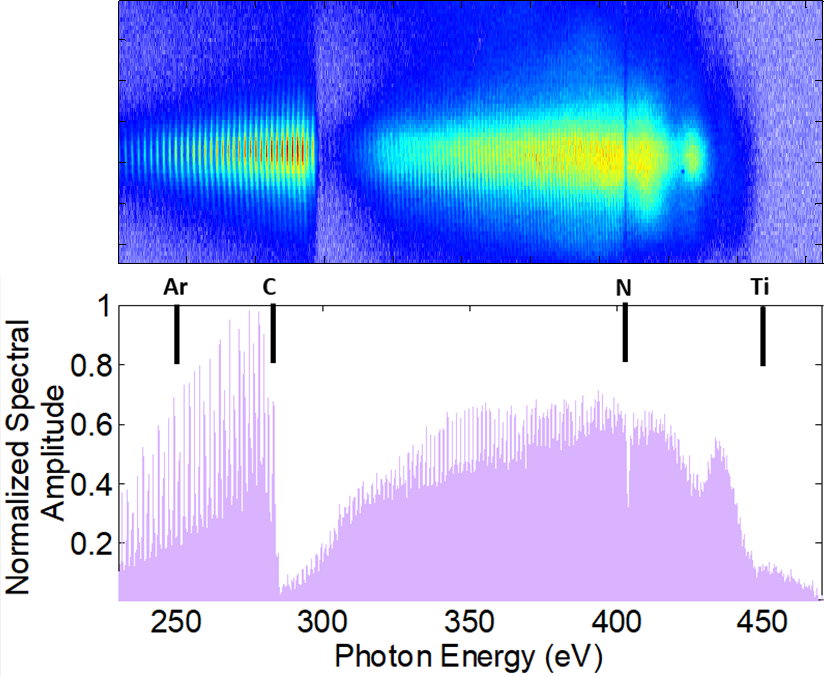
We are developing time-resolved soft x-ray absorption spectroscopy as a tool to investigate non-adiabatic processes in isolated few-body systems and in extended molecules. Using the element- and site-specificity of core shell transitions, we aim at visualizing ultrafast charge transfer processes in molecules. Dynamics at conical intersections, charge and energy transfer processes in push-pull chromophores and donor-acceptor molecules, and ligand-to-metal (et vice versa) charge transfer excited state dynamics in organometallic molecules are examples of the research directions. To this aim, we are developing sources of ultrashort soft x-ray pulse based on high-order harmonic generation. Additionally, experiments are performed at large scale facilities such as FLASH, the free electron laser in Hamburg, and the Linac Coherent Light Source (LCLS) at Stanford.
2.2 Ultrafast Molecular Dynamics
Project coordinators: A. Rouzée , K. AminiTopics
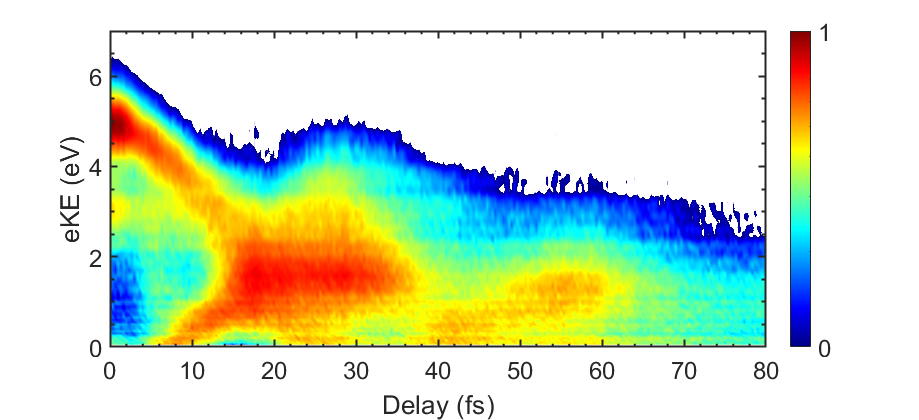
Over the past decades, femtosecond nonlinear spectroscopic techniques using optical and infrared laser pulses have established a new paradigm for observing and characterizing photo-induced structural changes in molecules and large biomolecular complexes. Here, we aim to extend these techniques into the VUV and XUV spectral ranges by employing state-of-the-art sources based on high-order harmonic generation and resonant dispersive-wave emission in hollow-core fibers. We aim to track charge flow in molecules on its natural time scale and to visualize how photo-induced electron dynamics couple to subsequent structural transformations.
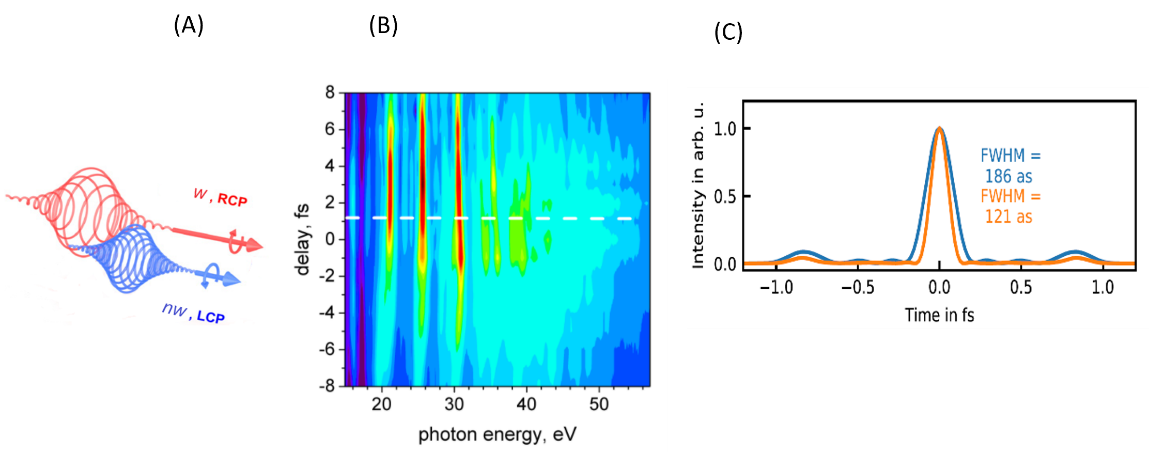
Part of Project 2.2 is dedicated to advancing the generation of attosecond pulses with controlled polarization. We have carried out an extensive series of studies on high-harmonic generation (HHG) dynamics in two-color, bi-circular (TCBC) driving schemes employing tailored fields with frequency ratios of ω/2ω, ω/3ω, and ω/4ω in the 400–1600 nm spectral range. These efforts culminated in the demonstration of a novel scheme for producing isolated attosecond pulses with high ellipticity, together with the full characterization of their polarization state (U. Bengs, N. Zhavoronkov, Scientific Reports11, 9570 (2021))

We use ultrafast electron diffraction (UED) to resolve structural changes in gas-phase molecules following photoexcitation with femtosecond temporal resolution and picometre spatial sensitivity. Elastic scattering of keV-MeV electrons provides direct access to transient nuclear geometries, enabling the visualization of bond rearrangements, vibrational wavepacket motion, and early-time structural signatures of nonadiabatic dynamics. In parallel, the detection of inelastically scattered electrons yields complementary information on excitation-induced electronic changes, offering additional insight into the coupling between electronic and nuclear motion. At MBI, we have a state-of-the-art, sub-100 fs 90 keV electron source operating at 40-100 kHz with direct electron detection capable of measuring both solid-state and gas-phase electron scattering. Moreover, by combining these UED measurements with complementary time-resolved spectroscopies such as transient absorption, our goal is to obtain a more complete description of photochemical reaction pathways in isolated molecules.