3.1 Dynamics of Condensed Phase Molecular Systems
Project coordinators: E. T. J. Nibbering , O. KornilovPhase 2 (2002-2006): Ultrafast dynamics of medium-strong intermolecular hydrogen bonds and of neat liquid water
The people involved in Phase 2:
Nils Huse, Karsten Heyne, Satoshi Ashihara, Agathe Espagne, Jens Dreyer, Erik T. J. Nibbering, Thomas Elsaesser
Collaboration within SFB-450 (Teilprojekt C1): Milena Petkovic,† Oliver Kühn†
†: Freie Universität Berlin, Institut für Chemie, Physikalische und Theoretische Chemie, Takustrasse 3, D-14195 Berlin, Germany.
International collaboration: Barry D. Bruner,* Michael L. Cowan,* Darren Kraemer,* Alexander Paarmann,* Jason R. Dwyer,* Brige Chugh,* Maher Harb,* R. J. Dwayne Miller ,* Shaul Mukamel#
*: Department of Chemistry and Physics, University of Toronto, 80 St. George Street, Toronto, Ontario, Canada, M5S 3H6.
#: Department of Chemistry, University of California at Irvine, Irvine, California, USA
This research project is embedded within the Collaborative Research Center
SFB 450 "Analysis and control of ultrafast photoinduced reactions"


Phase 2 (2002-2006):
In this period we investigated the vibrational couplings of a prototype medium-strong intermolecular hydrogen bonded system, acetic acid dimer, a model system that frequently appears in protein structures. Here the couplings of the O-H stretching vibration with the fingerprint O-H bending, and the C-O and C=O stretching modes, as well as with the low-frequency hydrogen bond modes have been explored. In addition we have been able to measure the dephasing, spectral diffusion and vibrational relaxation of the O-H stretching band of neat liquid water, the hydrogen bonded system with the fastest vibrational dynamics observed until now.
2-1 Coherent wavepacket motions in medium-strong intermolecular hydrogen bonds
2-2 Ultrafast dephasing, spectral diffusion and vibrational relaxation of neat liquid water
Coherent response in ordered medium strong hydrogen bonds
The generation of vibrational wave packets is an essential requisite for optical control of nuclear motion and requires the optical preparation of a coherent superposition of several nuclear wave functions. This should be achieved on hydrogen bonded systems in the electronic ground state through coherent excitation of several levels of low-frequency vibrational modes, that modulate the length of the hydrogen bond. We have indicated the potential feasibility of this coherent control scheme by excitation in the mid-infrared of the O-H or O-D stretching vibrational degrees of freedom, onto which the these low-frequency modes couple anharmonically. We show by use of femtosecond mid-infrared pump-probe and four-wave mixing spectroscopies that this anharmonic coupling can be determined in the time domain from which we learn the temporal characteristics (dephasing) of the vibrational wave packets, and the dephasing (T2) and population (T1) dynamics of the O-H/O-D stretching vibrations. From these nonlinear spectroscopic experiments we also aim to obtain information on the nature of the hydrogen bond by determination of the underlying direct (O-H/O-D stretch - solvent) and indirect (O-H/O-D stretch - low-frequency mode - solvent) coupling mechanisms that lead to dephasing and relaxation phenomena, the extraordinary characteristics of which also lead to the very specific line shapes of the O-H/O-D stretching modes, as exemplified by the strong red-shift, extreme broadening and peculiar substructures of these bands.
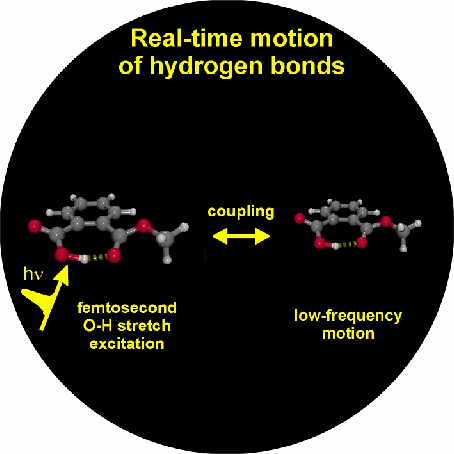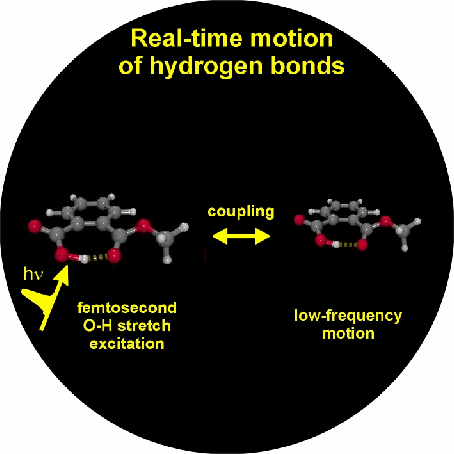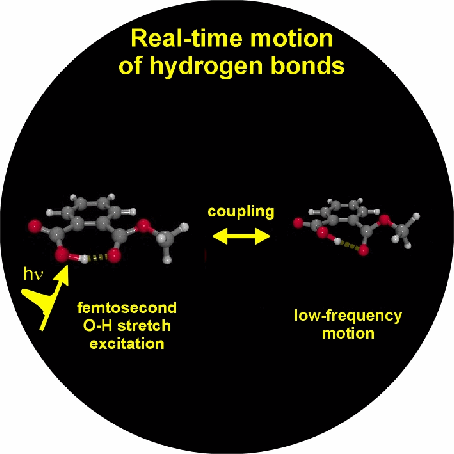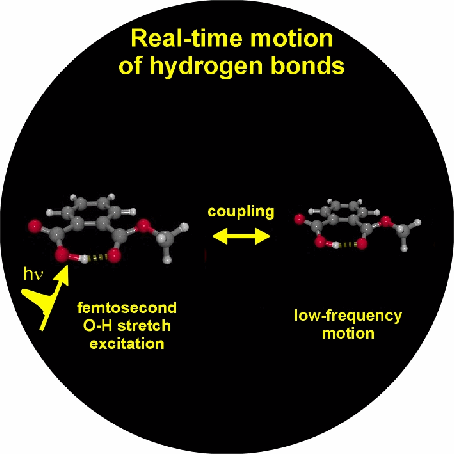
1. Vibrational perspective of hydrogen bonds: typically a strong anharmonic coupling exists between the O-H/O-D stretching and low-frequency modes that modulate the hydrogen bond distance. For this set of coupled oscillators this has the consequence that excitation of the O-H/O-D stretching mode includes excitation of these low-frequency modes, in a similar fashion as electronic excitation of molecules is accompanied by vibrational excitation. Ultrafast excitation of the O-H/O-D stretching mode in the mid-infrared thus leads to coherent wave packet motions of these low-frequency modes.
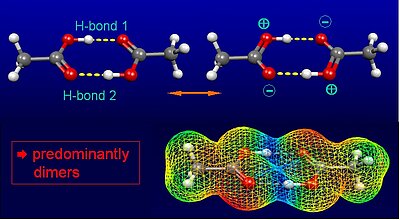
2. Acetic acid forms a cyclic dimer with two identical medium-strong hydrogen bonds in nonpolar solvents.
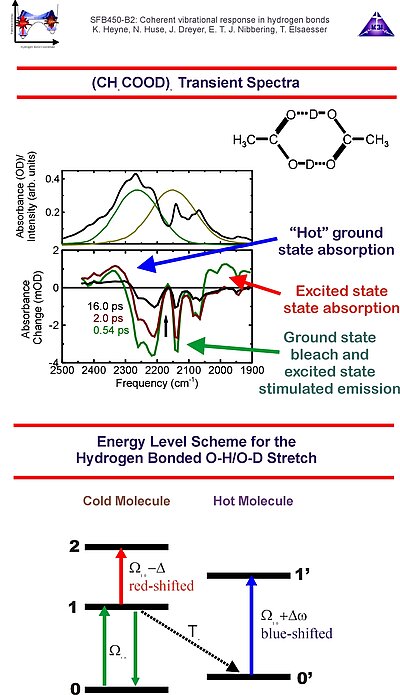
3. We have investigated several isotopomers of acetic acid dimer. The transient spectra can be explained by use of a a multi-level description for the O-H/O-D stretching mode. After excitation of the v=0 ® v=1 transition of the O-H/O-D stretching mode, one can detect absorbance changes due to v=0 ® v=1 bleach and v=1 ® v=0 stimulated emission, as well as v=1 ® v=2 excited state absorption, the latter is red-shifted due to anharmonicity of the O-H/O-D stretching mode. After population relaxation of the O-H/O-D stretching mode a hot molecule with a weaker hydrogen bond strength results, with a blue-shifted v'=0 ® v'=1 transition of the O-H/O-D stretching mode.
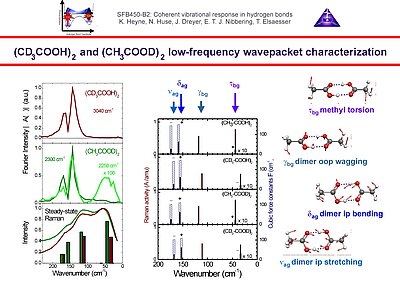
4. We have found modulations of pump-probe signals on the acetic acid dimer due to a weak contribution of the 50 cm-1 methyl torsion mode. Much stronger modulations, as evidenced by a beating pattern, are due to the in-plane bending and stretching modes with frequencies of 145 and 170 cm-1. The 115 cm-1 out-of-plane hydrogen bond deformation mode does not appear to contribute underdamped oscillations to the pump-probe signal. The experimental results are fully consistent with recent estimations of anharmonic couplings from ab initio quantem chemical calculations.
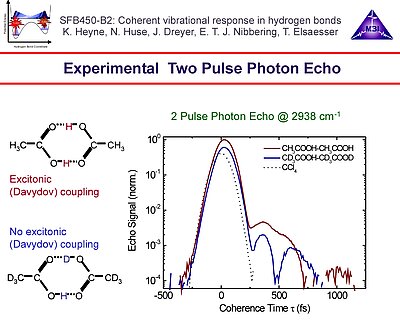
5. The intensity integrated two pulse photon echo of acetic acid dimer is dominated by quantum beats, caused by the multi-level substructure of the O-H stretching band. In particular the quantum beat contributions caused by the couplings with the low-frequency in-plane bending and in-plane stretching modes are prominent in these echo signals.
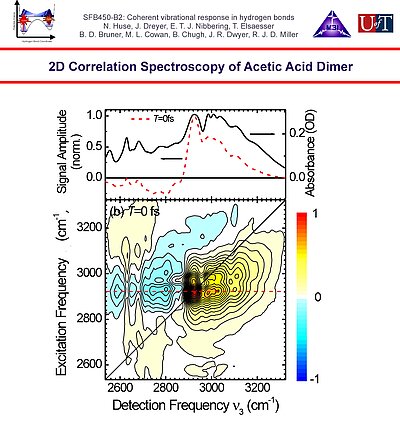
6. Heterodyne detected photon echo correlation spectroscopy of the O-H stretching band of acetic acid dimer reveals the significant mixing of the levels of the O-H stretching vibration with the combination overtone levels of the O-H and C-H bending, and the C-O and C=O stretching fingerprint modes through Fermi resonances.
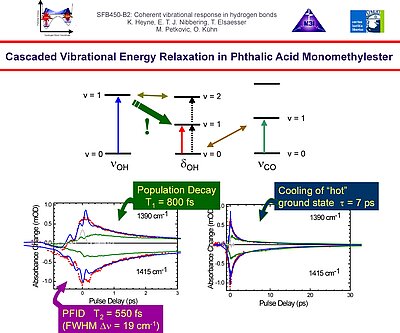
7. In phthalic acid monomethyl ester (PMME-H) we monitored with two-colour pump-probe spectroscopy the cascaded vibrational energy relaxation pathway that determines the fate of an O-H stretching excitation. We show that part of the excitation is channeled through the O-H bending vibrational levels.
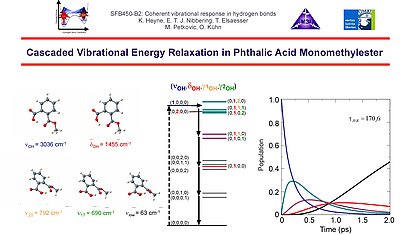
8. These experimental results on vibrational energy flow in PMME-H have been corroborated with quantum chemical calculations, that show besides the activity of the O-H bending the active role of two out-of-plane deformation modes.
Ultrafast dephasing, spectral diffusion and relaxation dynamics of the vibrational modes of neat liquid water
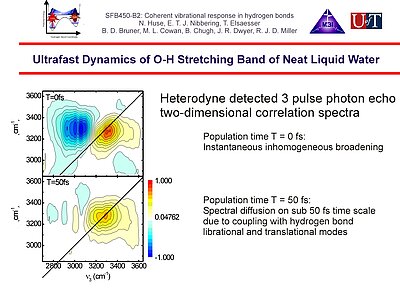
9. Heterodyne detected two-dimensional photon echo correlation spectroscopy of the O-H stretching band of neat liquid water shows an extremely fast spectral diffusion on a time scale of < 50 fs, that has to be ascribed to the impact of fluctuations through anharmonic coupling with librational modes.
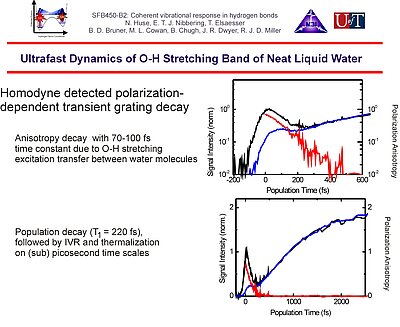
10. Polarization-sensitive transient grating spectroscopy of the O-H stretching band reveals a vibrational excitation transfer with a time constant of about 100 fs, whereas population relaxation only occurs with a T1 = 220 fs. Subsequent intramolecular vibrational redistribution and thermalization then proceeds on (sub)picosecond time scales.
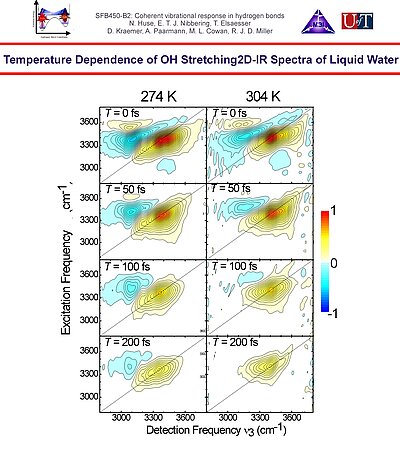
11. Temperature dependent 2D-IR spectra of the O-H stretching band of neat liquid water showing pronounced frequency dependent spectral diffusion dynamics at 274 K.
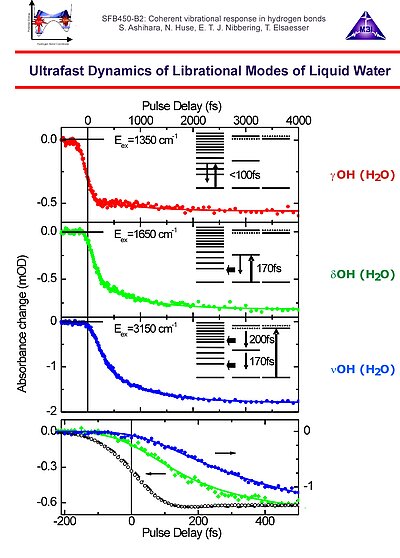
12. Two-colour pump-probe spectroscopy of the librational band of neat liquid water around 840 cm-1 showing dynamics dependent on the water mode being pumped: characteristic time scales < 100 fs for the water librational modes, T1 = 170 fs for the bending and T1 = 200 fs for the stretching mode relaxation. The delayed absorbance decrease in the case of bending and stretching excitation is shown in the lower panel, together with a cross correlaton measured in.a Ge substrate.