3.1 Dynamics of Condensed Phase Molecular Systems
Project coordinators: E. T. J. Nibbering , O. KornilovPhase 1 (1999-2002): Ultrafast dynamics of medium-strong intramolecular hydrogen bonds and of isotopically diluted water
The people involved in Phase 1:
J. Stenger, D. Madsen, J. Dreyer, E. T. J. Nibbering, P. Hamm, T. Elsaesser
This research project is embedded within the Collaborative Research Center
SFB 450 "Analysis and control of ultrafast photoinduced reactions"


Phase 1 (1999-2002):
Our research initally focussed on the investigation of the structurally well-defined medium-strong intramolecular hydrogen bonds of phthalic acid monomethyl ester and 2-(2'-hydroxyphenyl)benzothiazole, where we investigated the anharmonic couplings with low-frequency hydrogen bond modes, as well as the rapidly fluctuating hydrogen bond networks of isotopically diluted water (HOD in D2O).
1-1 Coherent wavepacket motions in medium-strong intramolecular hydrogen bonds
1-2 Ultrafast dephasing and spectral diffusion of isotopically diluted water
The generation of vibrational wave packets is an essential requisite for optical control of nuclear motion and requires the optical preparation of a coherent superposition of several nuclear wave functions. This should be achieved on hydrogen bonded systems in the electronic ground state through coherent excitation of several levels of low-frequency vibrational modes, that modulate the length of the hydrogen bond. We have indicated the potential feasibility of this coherent control scheme by excitation in the mid-infrared of the O-H or O-D stretching vibrational degrees of freedom, onto which the these low-frequency modes couple anharmonically. We show by use of femtosecond mid-infrared pump-probe and four-wave mixing spectroscopies that this anharmonic coupling can be determined in the time domain from which we learn the temporal characteristics (dephasing) of the vibrational wave packets, and the dephasing (T2) and population (T1) dynamics of the O-H/O-D stretching vibrations. From these nonlinear spectroscopic experiments we also aim to obtain information on the nature of the hydrogen bond by determination of the underlying direct (O-H/O-D stretch - solvent) and indirect (O-H/O-D stretch - low-frequency mode - solvent) coupling mechanisms that lead to dephasing and relaxation phenomena, the extraordinary characteristics of which also lead to the very specific line shapes of the O-H/O-D stretching modes, as exemplified by the strong red-shift, extreme broadening and peculiar substructures of these bands.
1-1 Coherent wavepacket motions in medium-strong intramolecular hydrogen bonds
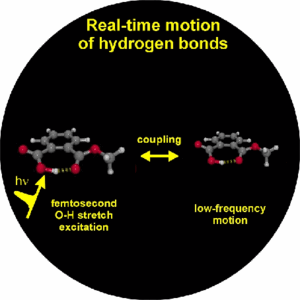
1. Vibrational perspective of hydrogen bonds: typically a strong anharmonic coupling exists between the O-H/O-D stretching and low-frequency modes that modulate the hydrogen bond distance. For this set of coupled oscillators this has the consequence that excitation of the O-H/O-D stretching mode includes excitation of these low-frequency modes, in a similar fashion as electronic excitation of molecules is accompanied by vibrational excitation. Ultrafast excitation of the O-H/O-D stretching mode in the mid-infrared thus leads to coherent wave packet motions of these low-frequency modes.
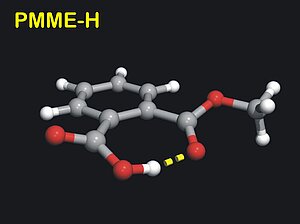
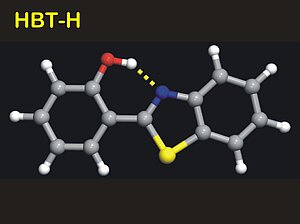
2. We have, for the first time, demonstrated this on organic molecules with a single intramolecular hydrogen bond, phthalic acid monomethyl ester (PMME) and 2-(2'-hydroxyphenyl)benzothiazole (HBT), in the ordinary forms (PMME-H) or in the deuterated (PMME-D or HBT-D) forms.
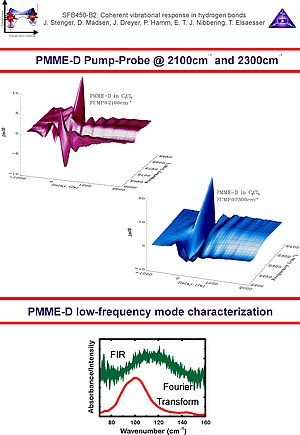
3. With femtosecond mid-infrared pump-probe spectroscopy we have been able to observe the low-frequency wave packet motions in PMME-H, PMME-D and HBT-D. Here, the oscillatory features in the pump-probe transients are compared with low-frequency steady-state far infrared and Raman spectra and normal mode calculations. In the case of PMME the wave packet motion is generated of a 100 cm-1 out-of-plane hydrogen bond deformation mode.
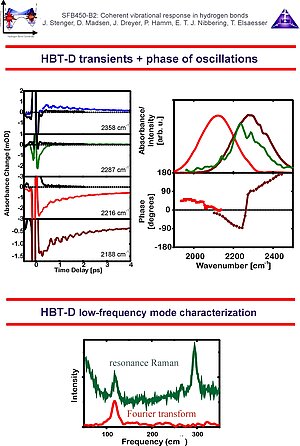
4. In the case of HBT-D the modulations are due to a 120 cm-1 in-plane hydrogen bond deformation mode.Since the T1 population relaxation time of the v=1 state of the O-D stretching mode is extremely short, the ground state wave packet motions dominate the pump-probe transients.The initial phase of the oscillations as function of probing frequency show a 180 degrees phase jump at the maximum of the absorption band, indicating a resonantly enhanced coherent excitation of the low-frequency mode.
1-2 Ultrafast dephasing and spectral diffusion of isotopically diluted water
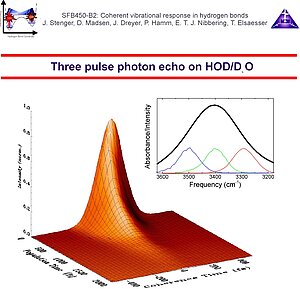
5. Hydrogen bonds are sensitive to the influences of the fluctuating environment. The solvent molecules can either directly couple to the O-H/O-D stretching oscillators (direct coupling mechanism) or they can perturb the motions of the low-frequency hydrogen bond modes, and, since the latter are anharmonically coupled to the O-H/O-D stretching modes, a coherence loss results (indirect coupling mechanism). Either way, the O-H/O-D stretching polarization decays, and accordingly the transitions are broadened. Compared to typical infrared transitions, the broadening is extreme, thereby masking any fine details. The question whether the line broadening is either homogeneous (due to a fast fluctuating environment) or inhomogeneous (due to a static distribution of hydrogen bond distances and angles), or that both contribute. An even better question is which characteristic time scales the O-H/O-D stretching frequency fluctuation correlation function has. With this quantity it is possible to estimate the relative contributions of homogeneous dephasing, spectral diffusion and inhomogeneous broadening. The method to determine this frequency fluctuation correlation function is femtosecond infrared photon echo spectroscopy. We have performed photon echo experiments at 3 different frequency positions in the O-H stretching band of HOD in D2O.
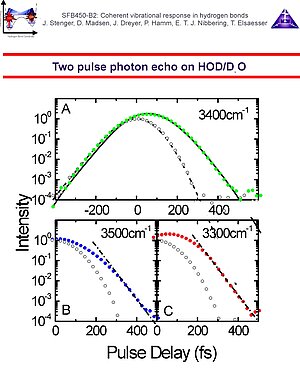
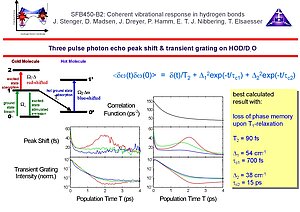
6. We have found that the two pulse photon echo decay is determined by an extremely short dephasing time, a consequence of the combined effect of a rapid fluctuating environment and the extreme anharmonicity of the O-H/O-D stretching oscillator. With our time resolution of 130 fs we have not been able to distinguish between the assumption of an infinitely short correlation time (justifying the use of the dephasing time constant T2 = 90 fs) or a Kubo-Ansatz where the fits converged to a correlation time of about 30 fs. In any case 50% the line width is given by this fast dephasing component. Three pulse echo peak shift measurements indicate that in addition to this rapid component of the correlation function, additional dynamics occurs with 700 fs and 15 ps time constants. To describe the echo peak shift measurements correctly it is important to take into account the multilevel structure of the O-H/O-D stretching modes, where the v=1 state decays into a "hot" ground state with T1 = 700 fs without conservation of phase memory. The bottom line in these experiments is that phase memory O-H/O-D stretching bands in intermolecular hydrogen bonds with a large geometric flexibility (such as in water) decays on a multitude of time scales. The consequence is that structural features (distribution of hydrogen bond lengths/angles etc.) persist on relatively long times.
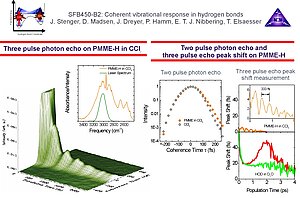
7. We have performed femtosecond mid-infrared four wave mixing experiments on the O-H stretching mode in phthalic acid monomethyl ester (PMME-H) in tetrachloromethane. We have found that in contrast to HOD/D2O the line broadening in PMME-H is dominated by a homogeneous ultrafast dephasing process, without any spectral diffusion occuring on longer time scales, reflecting the well-defined geometry of the intramolecular hydrogen bond in PMME-H. The three pulse echo peak shift is basically negligible in magnitude compared to that in HOD/D2O. The anharmonic coupling with the underdamped 100 cm-1 mode leads to a modulation of the echo peak shift signal.