1.1 Fundamentals of Extreme Photonics
Project coordinators: O. Smirnova , K. BuschT5: Condensed matter theory
Ultra fast spin and charge dynamics

Ultrafast manipulation of spins in a controlled manner is a milestone of solid state physics. The motivation for this is to use electronic spin for storing binary data, which can then be optically manipulated using lasers. The advantage of such a technique would be reduction in the size and efficiency of the storage device by several orders of magnitude. Recent experiments have demonstrated that demagnetization or spin- reorientation processes can be induced by femtosecond laser pulses. However, we are still far from achieving optimally controlled manipulation of spins required for production of devices. One of the reasons behind this is the lack of full understanding of the phenomena leading to demagnetization. Time-dependent density functional theory (TDDFT) is a formally exact method for describing the real-time dynamics of electrons under the influence of an external field.
We use spin-resolved TDDFT to study of the ultrafast physics of
- Optical inter-site spin transfer
- Optical switching
- Demagnetization
Linear response
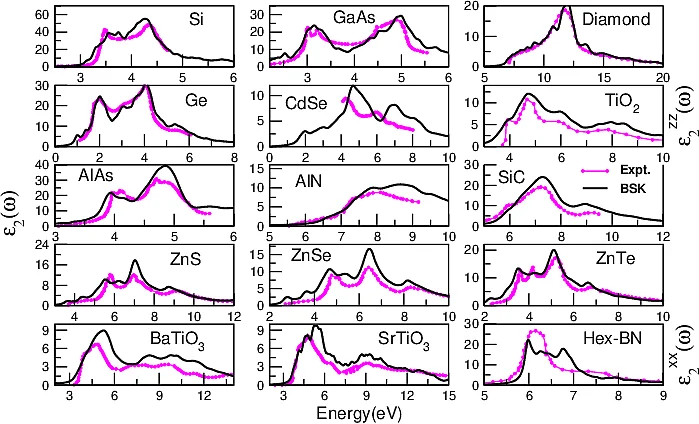
A complete understanding of a material requires the knowledge of the excited states of the system. In particular, the low energy excitations are of utmost importance while studying the electronic, optical magnetic, dynamical, and thermodynamical properties of the material. Time-Dependent Density Functional Theory (TD-DFT), within the linear response regime, is a successful and fully ab-initio method to access the electronic charge and spin excitations. Using TD-DFT we study charge-charge and spin-spin response function to accurately describe:
- Excitons
- Magnons
- Magnetic circular and linear dichroism (MCD/MLD)
Strong correlations
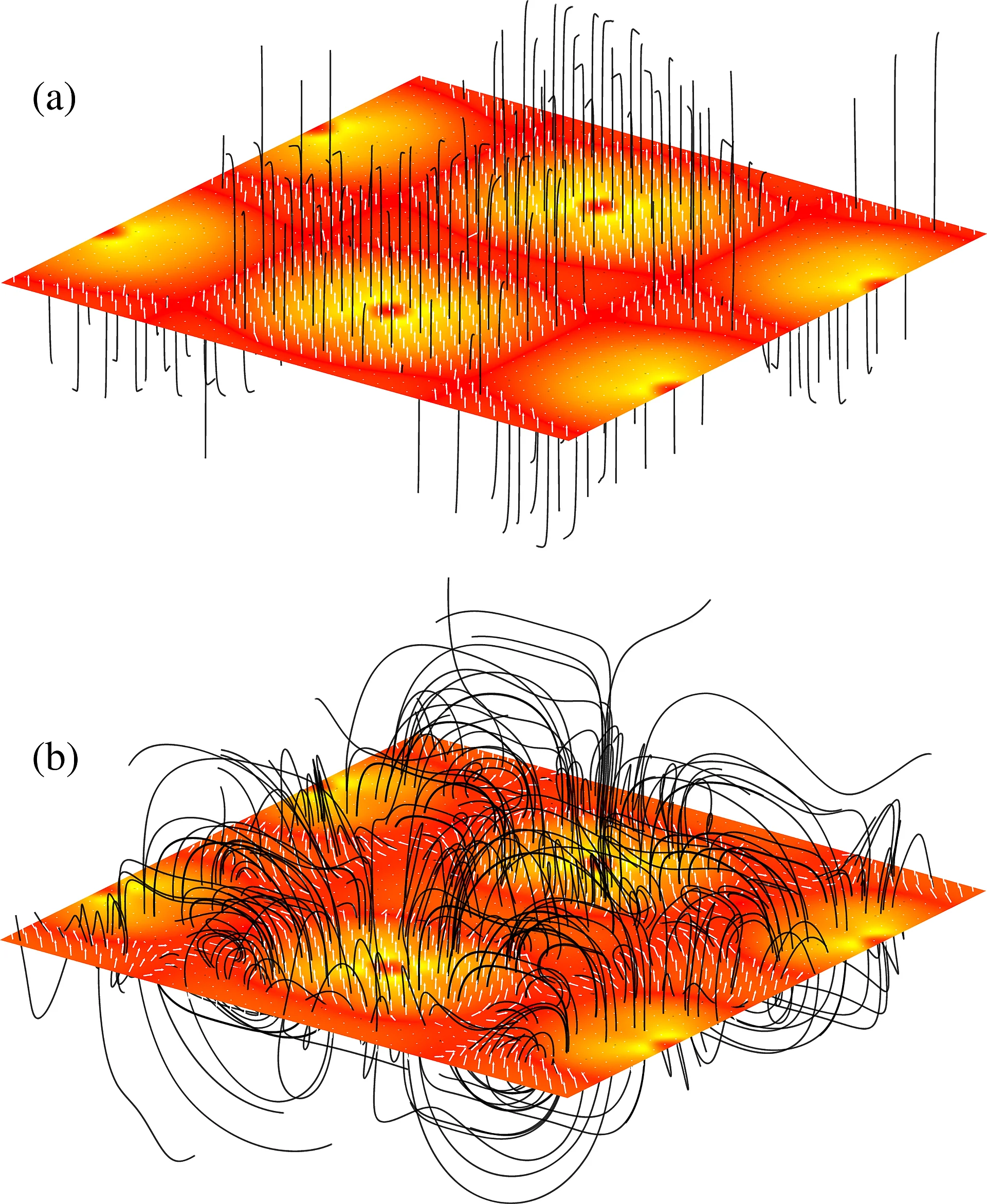
One of the most dramatic failures of the usual local density approximation or generalized gradient type approximations to the exchange-correlation functional of density functional theory is the incorrect prediction of a metallic ground state for the strongly correlated Mott insulators. A real challenge for any kind of ab-initio theory is the prediction of an insulating state for strongly correlated materials in the absence of magnetic order. To achieve this We have extended reduced density matrix functional theory (RDMFT) for the case of solids and have designed a new exchange-correlation functional. Using this we are able to accurately describe:
- Band gaps in Mott insulators
- Spectral function for Mott insulators
- Ionization energies and electron affinities for finite systems
Exachange correlation functionals within density functional theory
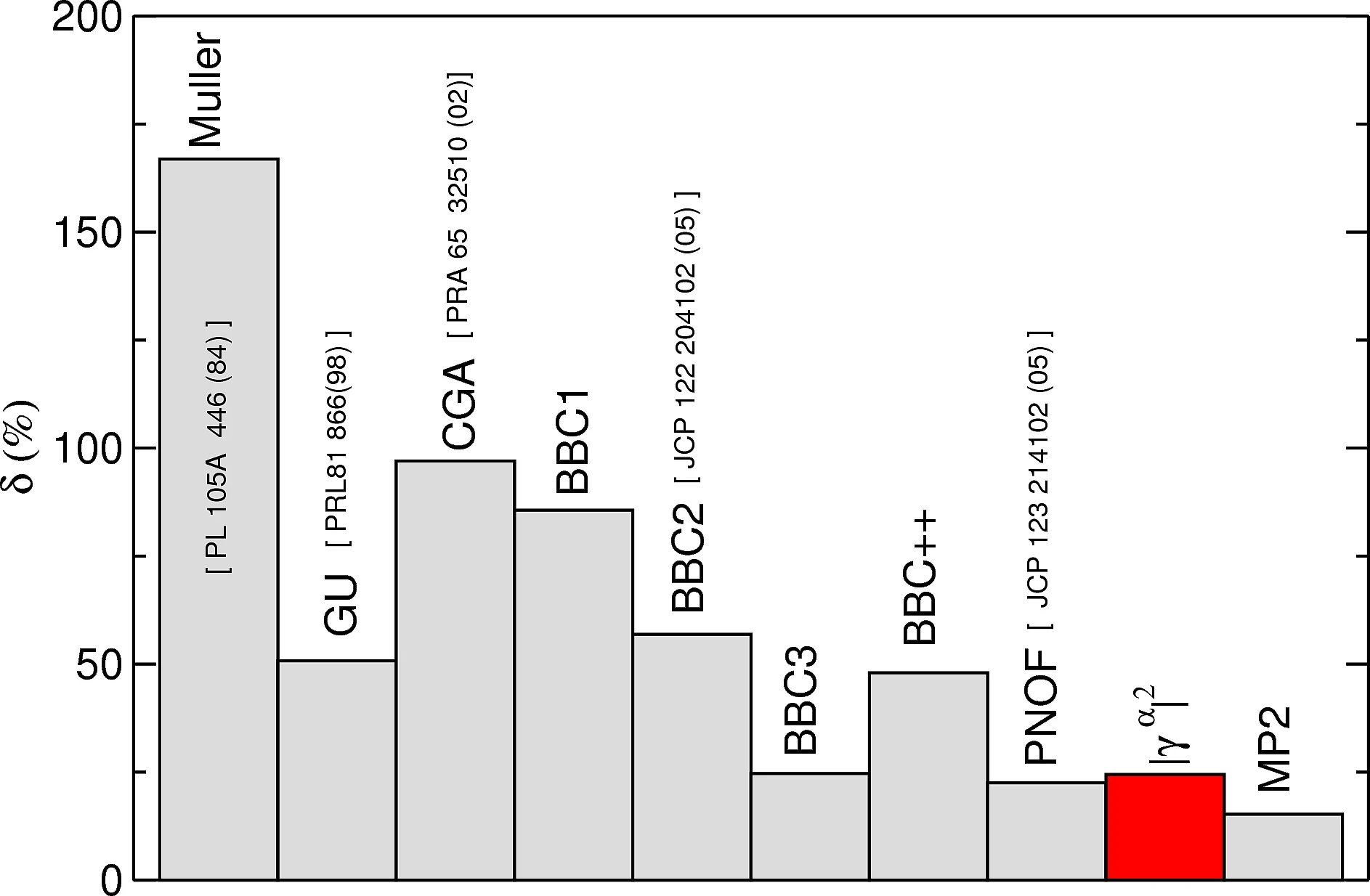
Density functional theory (DFT) and its extension to time domain, i.e. time-dependent density functional theory (TD-DFT) are in principle exact theories to calculate the ground-state and excited state properties respectively of any material. Although formally exact, the predictions of these functional theories are only as good as the approximation of the exchange-correlation (xc) potential and kernel. By designing new functionals we more accurately describe excitons and magnetic ground-states.
- Bootstrap functional for excitons in solids
- Source free functional for magnetic solids
Superconductors
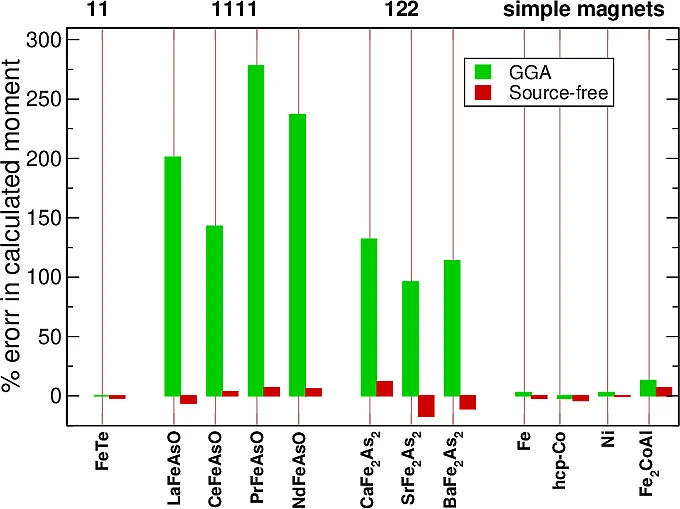
Density functional theory (DFT) has proven enormously successful for calculating the electronic structure of both molecules and solids. Lattice structures, phonon spectra and many other properties are now routinely calculated. Magnetism presents more of a mixed picture. Simple magnets, such as elemental solids (Fe, Co and Ni), are well-described by at least as far as total moments are concerned. However, all existing exchange-correlation functionals fail to describe the iron pnictide and related materials for which they greatly overestimate the moments by factors of two or more. We design a new source-free functional within DFT, which is able to accurately describe the normal state properties of superconducting pnictides, giving us hope to be able to in future correctly describe phase transition in these materials.
- Pnictides
Graphene
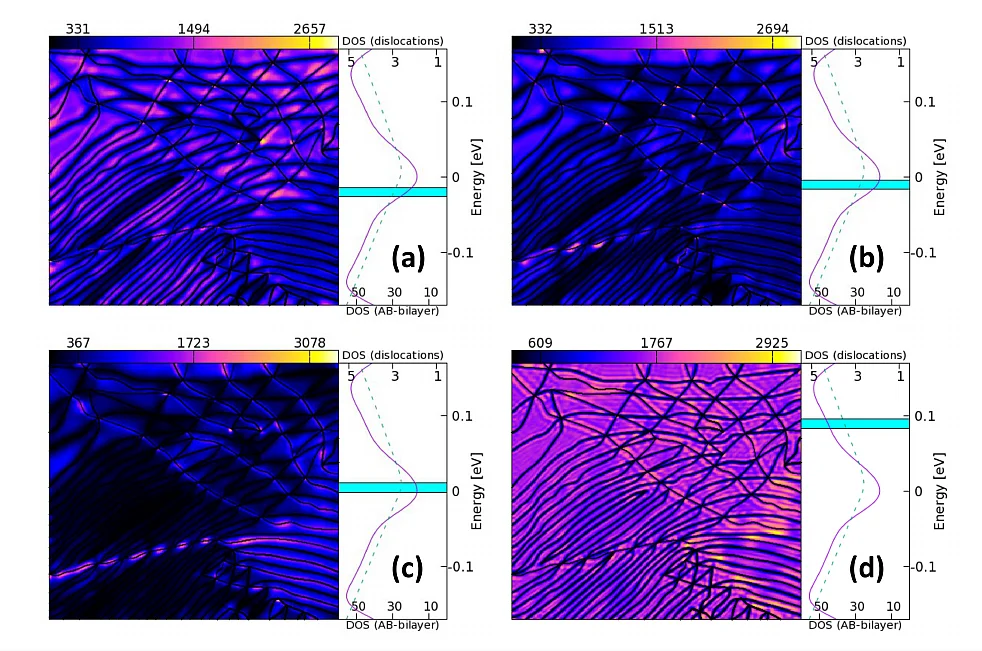
Deformations in materials are either perturbative or non-perturbative departures from the high symmetry crystal. Long wavelength ripples in a two-dimensional (2d) membranes belong to the former class, while recently imaged partial dislocations in bilayer graphene belong to the latter class. We design a theoretical approach capable of treating both perturbative and non-perturbative deformations on an equal footing. Using this theory we show why charge transport at the Dirac point in graphene exhibits two dramatically different transport states, insulating and metallic, that occur in apparently otherwise indistinguishable experimental samples.