Spectroscopic Probes of Non-Adiabatic Relaxation Dynamics
Crossings of electronic potential energy surfaces in nuclear configuration space, known as conical intersections, determine the rates and outcomes of a large class of photochemical molecular processes. Much theoretical progress has been made in computing strongly coupled electronic and nuclear motions at different levels, but how to incorporate them in different spectroscopic signals and the approximations involved are less established. We have established a wide range of time-resolved spectroscopic techniques which span from the infrared to the X-ray regimes and can be used for probing the nonadiabatic dynamics in the vicinity of conical intersections.
Transient electronic and vibrational probes and their theoretical signal calculations are classified by their information content. This includes transient vibrational spectroscopic methods (transient infrared and femtosecond off-resonant stimulated Raman), resonant electronic probes (transient absorption and 2D UV/Vis spectroscopy), and novel stimulated X-ray Raman techniques.
Along with the precise definition of what to calculate for predicting the various signals, a toolbox of protocols for their simulation is outlined [for details see links below and a recent Review: Chem. Rev., 117, 12165 (2017)].
- Femtosecond Stimulated Raman Spectroscopy of the DNA Photoprotection Mechanism[J. Chem. Theory Comput. 10, 1172 (2014)]:
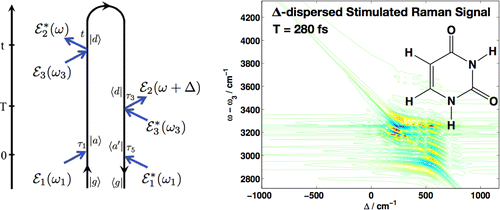
Nonadiabatic electron and nuclear dynamics of photoexcited molecules involving conical intersections is of fundamental importance in many reactions such as the self-protection mechanism of DNA and RNA bases against UV irradiation. Nonlinear vibrational spectroscopy can provide an ultrafast sensitive probe for these processes. We employ a simulation protocol that combines nonadiabatic on-the-fly molecular dynamics with a mode-tracking algorithm for the simulation of femtosecond stimulated Raman spectroscopy (SRS) signals.
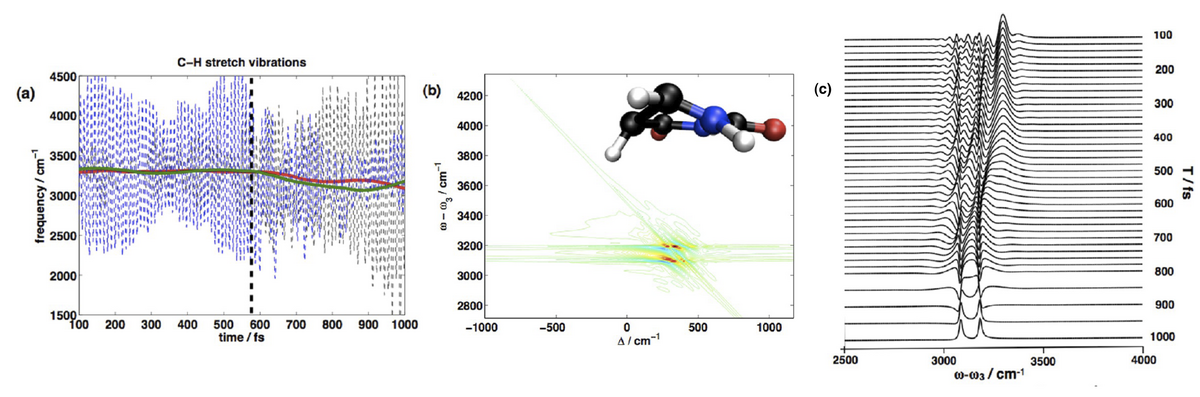
Analysis of the joint time/frequency resolution of the technique reveals a matter chirp contribution that limits the inherent temporal resolution and characteristic signatures of relaxation dynamics mediated in the vicinity of conical intersection are predicted. Employed localized vibrations provide high sensitivity to their local environment and act as local probes with submolecular and high temporal resolution.
- Nonlinear 2D spectra of molecular systems in the UV-Vis spectral domain [J. Chem. Theory Comput., 12, 3284 (2016)]:
We develope an algorithm for the simulation of nonlinear 2D spectra of molecular systems in the UV-Vis spectral region from atomistic molecular dynamics trajectories subject to non-adiabatic relaxation. To achieve this, we combine the nonlinear exciton propagation protocol, that relies on a quasiparticle approach with the surface hopping methodology to account for quantum-classical feedback during the dynamics. Phenomena like dynamic Stokes shift due to nuclear relaxation, spectral diffusion and population transfer among electronic states are thus naturally included. The algorithm can be applied to a variety of systems ranging from simple two-state models to complex organic molecules like the bichromophore diphenylmethane.
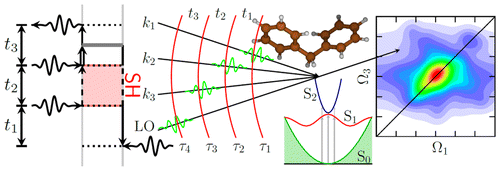
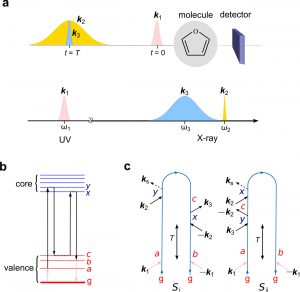
- Attosecond Stimulated X-ray Raman Spectroscopy the Ring Opening of Furan[Structural Dynamics 3, 023601 (2016)]:
Attosecond X-ray pulses are short enough to capture snapshots of molecules undergoing nonadiabatic electron and nuclear dynamics at conical intersections (CoIns). A stimulated Raman probe induced by a combination of an attosecond and a femtosecond pulse has a unique temporal and spectral resolution for probing the nonadiabatic dynamics and detecting the ultrafast (~ 4.5 fs) passage through a CoIn. This is demonstrated by a multiconfigurational self-consistent-field study of the dynamics and spectroscopy of the furan ring-opening reaction. The signals are highly sensitive to the changes in nonadiabatically coupled electronic structure and geometry.